Pratiche di ispezione visiva delle apparecchiature pulite
Secondo le linee guida normative e gli standard di farmacopea, i produttori devono confermare che le apparecchiature di processo siano visivamente pulite dopo un intervento di pulizia. Di recente, un'indagine Steris ha dimostrato che le pratiche di ispezione visiva delle attrezzature pulite variano da un'azienda all'altra, così come il fatto che, nonostante le pratiche e perfino la terminologia possano differire, ciò può essere accettato dagli enti regolatori a condizione che i processi siano ben documentati.
2020-05-12
Secondo le linee guida normative e gli standard di farmacopea, i produttori devono confermare che le apparecchiature di processo siano visivamente pulite dopo un intervento di pulizia. Di recente, un'indagine Steris ha dimostrato che le pratiche di ispezione visiva delle attrezzature pulite variano da un'azienda all'altra, così come il fatto che, nonostante le pratiche e perfino la terminologia possano differire, ciò può essere accettato dagli enti regolatori a condizione che i processi siano ben documentati.
Innanzitutto, una parola sulla terminologia: molte linee guida normative e documenti tecnici del settore usano termini differenti per riferirsi alla fase che conferma che l'apparecchiatura di processo è visivamente pulita dopo la pulizia. Ai fini del presente articolo, gli autori usano il termine “ispezione visiva”. Altri termini comuni per questa fase sono “controllo visivo” o “esame visivo”. Si noti che in questo caso, “ispezione visiva” non si riferisce al controllo visivo del prodotto finito per individuare eventuali particelle.
Hanno risposto al sondaggio 39 aziende in rappresentanza di numerose aziende farmaceutiche e biofarmaceutiche europee (27 aziende in 34 sedi differenti). I produttori che hanno completato il sondaggio operano nei seguenti ambiti: 54% non sterile (ad es. compresse, liquidi, prodotti combinati), 13% sterile (ad es. biotecnologie, prodotti liquidi e liofilizzati), 26% vaccini, 5% dispositivi medici, 2% altro (ad es. produzione clinica iniziale).
L’allegato 15 delle GMP europee dichiara che “Il controllo visivo per la pulizia è un elemento importante dei criteri di accettazione per la convalida della pulizia” (1). L'ispezione visiva è una fase fondamentale per confermare l'efficacia del processo di pulizia dell'apparecchiatura dopo la pulizia. Il criterio di accettazione per l'ispezione visiva è visivamente pulito. L'ispezione visiva deve includere le superfici a contatto diretto e indiretto con il prodotto e richiede che le superfici dell'apparecchiatura siano visibili. In caso contrario, potrebbe essere necessario smontare l'apparecchiatura per avervi accesso o utilizzare strumenti come specchi, fonti luminose o endoscopi (2, 3). Per recipienti di grandi dimensioni, si possono prendere in considerazione tecnologie di ultima generazione, come fotocamere digitali in grado di valutare la superficie, quando l’ispezione visiva risulta difficoltosa.
Visivamente pulito è lo standard minimo previsto. Tuttavia, viene imposto un ulteriore criterio di accettazione, come il limite di sicurezza per la salute (1-5). Pertanto, un metodo analitico convalidato con una sensibilità inferiore al limite di pulizia deve essere periodicamente abbinato a un'ispezione visiva. Quando si applica solo l'ispezione visiva per determinare la pulizia dell'apparecchiatura, è necessario stabilire la soglia alla quale il prodotto è facilmente visibile sotto forma di residuo (3, 6).
L'ispezione visiva si esegue sempre (nei limiti del possibile) al termine di un ciclo di pulizia completo (7). L'ispezione visiva è un'osservazione attiva e qualitativa delle superfici a contatto con il prodotto per confermare che non sono presenti residui e che si può iniziare la produzione del lotto successivo (7). ICH Q7: Le Linee guida pratiche per gli ingredienti farmaceutici attivi riportano quanto segue: “12,76… L’ispezione visiva può consentire il rilevamento di contaminazioni grossolane concentrate in piccole aree che altrimenti potrebbero non essere rilevate con il campionamento e/o con l’analisi.”
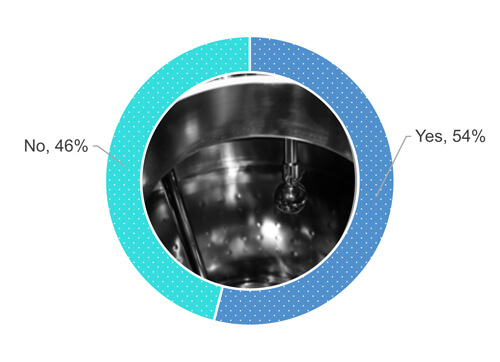
Tornando all'indagine, il 54% degli intervistati esegue un'ispezione visiva della superficie dell'apparecchiatura quando è asciutta (Figura 1).
Figura 1 La procedura aziendale prevede l'ispezione visiva della superficie dell'apparecchiatura di processo dopo un'operazione di pulizia, quando la superficie è asciutta?
Alcuni documenti tecnici del settore suggeriscono che l'ispezione visiva deve essere eseguita su una superficie asciutta, quando possibile, per evitare risultati falsi negativi (8,9):
Linee guida del Comitato per i principi attivi farmaceutici (10):
“Dopo aver eseguito le procedure di pulizia, l’apparecchiatura deve essere asciugata per consentirne l’ispezione visiva.”
“I criteri di accettazione per la pulizia delle apparecchiature devono basarsi su una pulizia visibile in condizioni asciutte e su un limite analitico.”
Relazione tecnica della PDA n. 29: Aspetti da considerare per la convalida della pulizia (9): “Normalmente le superfici sottoposte a ispezione visiva devono essere asciutte, in quanto tale condizione rappresenta lo scenario più sfavorevole per l’ispezione stessa.”
È noto che, per alcuni residui, la pulizia visibile si ottiene solo quando la superficie è bagnata, mentre ciò non avviene più quando la superficie è asciutta.
La configurazione dell'apparecchiatura e i parametri del ciclo di pulizia possono contribuire ad asciugare le superfici dell'apparecchiatura, ad esempio disponendo le tubazioni in pendenza verso lo scarico, rendendole autodrenanti ed effettuando l’ultimo risciacquo ad alte temperature. In alcuni casi, l'aria pulita soffiata nell'apparecchiatura e nel sistema di distribuzione può aiutare ad asciugare le superfici (9).
Nonostante il rispetto dei parametri sopra elencati, è possibile che sulla superficie siano presenti goccioline o umidità (“sudore”), che potrebbero essere accettabili se adeguatamente giustificate.
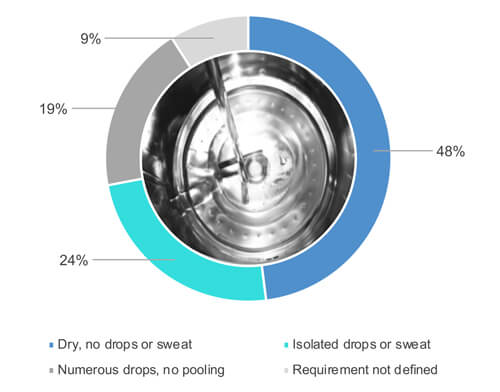
Come mostra la Figura 1, il 48% del 54% di coloro che hanno risposto di eseguire l’ispezione visiva delle superfici delle apparecchiature lo fa quando sono completamente asciutte (Figura 2). Il 24% del 54% accetta una certa umidità parziale (ad esempio gocce isolate o sudore) delle superfici durante un'ispezione visiva dell'apparecchiatura pulita (Figura 2). Il 19% del 54% accetta la presenza di un elevato numero di gocce, ma non di ristagno di acqua. Il 9% del 54% non ha definito i requisiti di asciuttezza.
Figura 2 Qual è il requisito di asciuttezza per l'apparecchiatura di processo al termine del ciclo di pulizia?
Sono accettabili diversi livelli di asciuttezza se adeguatamente giustificati e supportati da dati che dimostrino il non impatto sull'ispezione visiva post-pulizia e sulla proliferazione della carica batterica durante lo stoccaggio pulito, ad esempio tramite istruzioni e formazione approfondite mediante foto per evitare deviazioni. Infine, nessuna azienda sul 54% di quelle che ispezionano visivamente le apparecchiature quando le superfici sono asciutte autorizza il ristagno o l’accumulo di acqua sulle superfici delle apparecchiature, come suggerito nella guida per le ispezioni della FDA (10): “…Ad esempio, l’apparecchiatura deve essere asciugata prima dello stoccaggio e non deve mai essere presente un ristagno d’acqua dopo le operazioni di pulizia”.
Sta all'utente stabilire se le superfici delle apparecchiature sono visivamente pulite. Pertanto, gli utenti devono essere in grado di ispezionare visivamente tutte le superfici dell'apparecchiatura. Se non è possibile, devono essere disponibili strumenti adeguati o avanzati per garantire un corretto processo decisionale (2, 3, 9, 11). L'ispezione visiva di un recipiente di grandi dimensioni attraverso un vetro spia è restrittiva per via della superficie nascosta alla vista. L’EMA suggerisce che la capacità di ispezionare visivamente l’apparecchiatura, come le distanze osservate sul campo, deve essere presa in considerazione in un documento di domande e risposte (3), raccomandando che "devono essere disponibili istruzioni scritte che specifichino tutte le aree che richiedono un’ispezione visiva e i registri dovrebbero confermare chiaramente che tutte le ispezioni sono state completate". Infine, per garantire un processo decisionale corretto, sono obbligatorie procedure dettagliate e una formazione sui criteri visivi di pulizia. Il livello di formazione e qualificazione per l'ispezione visiva deve essere commisurato al rischio di contaminazione incrociata (3, 7, 9, 11).
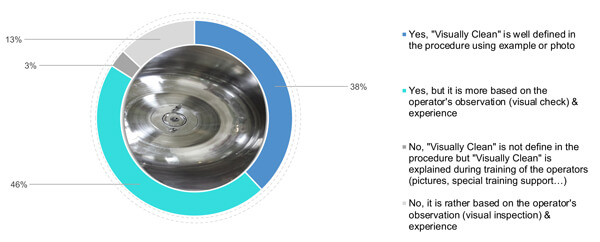
Per quanto riguarda le foto, il 38% degli intervistati ha definito criteri visivi di pulizia allegando anche fotografie ed esempi di uno stato pulito (Figura 3). Il 46% si affida alla comprensione del concetto di visivamente pulito dell'utente, abbinata a una definizione teorica di visivamente pulito nella procedura. Infine, il 16% (3% e 13%) si affida all'esperienza e alla formazione dell'utente (utilizzando un supporto specifico o immagini) che deve valutare la pulizia visibile di una superficie (Figura 3).
Figura 3 La vostra azienda definisce e descrive in dettaglio il significato dei criteri di "visivamente pulito"?
Gli utenti che eseguono l'ispezione visiva necessitano di una formazione specifica perché ciò che si può vedere visivamente varia a seconda della distanza, dell'angolazione, dell'illuminazione, della natura della superficie, del livello di asciugatura e dell'acuità visiva dell'esaminatore (3, 7, 9). L'EMA suggerisce che il test della vista (o dell'acuità visiva) dovrebbe essere eseguito periodicamente e che la competenza dell'utente dovrebbe essere dimostrata attraverso una valutazione pratica (3). La ISPE Risk Mapp suggerisce che: "Nelle situazioni in cui il metodo di rilevamento è esclusivamente visivo, è importante comprendere l'acuità visiva del personale e quale livello di residuo è considerato sicuro (7). Se il livello di sicurezza è al di sotto dell'acuità visiva del personale, il rischio che un problema non venga rilevato può essere considerato elevato, mentre se il livello di sicurezza è ben al di sopra (di diversi ordini di grandezza) dell'acuità visiva del personale, il rischio che il problema non venga rilevato può essere considerato basso.
La frequenza del test dell'acuità visiva e il limite dell'acuità visiva dipenderebbero da variabili specifiche come:
la distanza tra l'esaminatore e la superficie dell'apparecchiatura ispezionata (3, 7, 12)
la configurazione dell'apparecchiatura e la natura della superficie (7,8)
le condizioni di illuminazione dell'ambiente (3, 7, 9, 12)
la frequenza dei test analitici (3, 7-9, 11,12)
la tossicità dei residui, il limite di pulizia rispetto al limite di rilevamento visivo (3,6,7)
l'ispezione visiva, eseguita solo per confermare la pulizia di un'apparecchiatura (7)
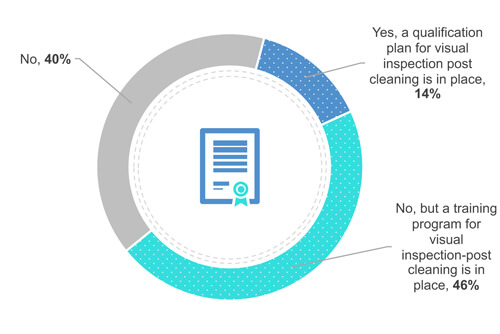
Il 38% degli intervistati non richiede alcuna certificazione o qualificazione dell'utente (Figura 4). Tuttavia, il 49% forma i propri utenti sul campo (ossia davanti all'apparecchiatura) in materia di pulizia visibile. Infine, il 13% esegue la qualificazione dei propri utenti.
Figura 4 La vostra azienda dispone di un programma di certificazione o qualificazione per l'ispezione visiva?
Alcuni produttori hanno definito tramite provini il limite visivo dei residui rilevabili da un utente (13, 14). Inoltre, alcuni di essi hanno imitato la distanza tra l'utente e le superfici da ispezionare sulle apparecchiature di processo (15, 16). Quando il limite visivo dei residui è inferiore al limite di pulizia, l'ispezione visiva da sola può essere sufficiente come criterio di accettazione (7).
Una certificazione o una qualificazione, insieme a un programma di formazione, garantiscono la competenza degli utenti nell'ispezione visiva di una superficie? Seguendo il precetto dell'ICH Q9: Gestione del rischio di qualità (17), la risposta dipenderebbe generalmente da:
numero di deviazioni storiche dell'ispezione visiva dovute alla formazione dell'utente
difficoltà nell'ispezione dell'apparecchiatura (configurazione dell'apparecchiatura e condizioni dell'ambiente circostante come illuminazione, angolazione, ecc.) (3, 7–9, 11, 12, 18, 19)
tipo di ispezione visiva utilizzata; ispezione visiva qualitativa o quantitativa (7,12, 13–16, 18,19)
ispezione visiva non integrata da test analitici (13–16,18)
frequenza dei test analitici eseguiti con l'ispezione visiva
sensibilità del metodo analitico rispetto a limite di pulizia, tossicità (sulla base del limite di esposizione alla salute) dei residui (1–7, 9,11)
presenza di un doppio controllo
Conclusione
Il concetto di ispezione visiva e i criteri di "visivamente pulito" sembrano variare tra i produttori europei in base alla loro esperienza nell'esecuzione del processo di pulizia e alla comprensione dei requisiti normativi. I criteri di pulizia visibile devono tuttavia essere chiaramente definiti nelle procedure. Gli utenti che eseguono ispezioni visive necessitano di una formazione specifica, che può basarsi sulle loro esperienze personali.
La variabilità nelle pratiche tra produttori, come suggerito dai risultati dell'indagine, può essere accettabile quando il rischio è documentato. Seguendo i precetti dell'ICH Q9, il livello di impegno e formalità deve essere commisurato al rischio del paziente.
Nella Parte II verrà presentato un case study e verranno suggeriti i requisiti minimi per l'ispezione visiva delle superfici pulite.
Parte II
I produttori europei interpretano i criteri di "visivamente pulito" in vari modi, sulla base della propria esperienza nell’esecuzione del processo di pulizia e delle proprie interpretazioni dei requisiti normativi (1). Tuttavia, è necessario stabilire un programma formale di formazione e qualificazione per l'ispezione visiva e le relative istruzioni per prevenire il rischio di contaminazione e per verificare che l'apparecchiatura venga pulita in base a una serie di controlli del produttore (2).
La variabilità nelle pratiche tra produttori è accettabile. Tuttavia, gli utenti devono essere formati e qualificati periodicamente su quando ispezionare visivamente le apparecchiature pulite e su come distinguere tra residui di prodotto e imperfezioni superficiali che potrebbero influire sui criteri di pulizia visibile. Il case study riportato di seguito illustra l'importanza di tali premesse.
Case study: osservazione dell'ispezione durante l'ispezione visiva
Di recente, gli ispettori dell'FDA statunitense hanno trasmesso a un produttore biofarmaceutico anonimo un'osservazione in merito ai criteri di pulizia visibile. Durante una visita al reparto produttivo dell'azienda, uno degli ispettori ha ispezionato la stanza a monte, nella quale erano posizionati due fermentatori. I recipienti fanno parte di un sistema chiuso e vengono puliti utilizzando un sistema CIP (clean-in-place) completamente automatizzato. Il ciclo CIP di uno dei fermentatori era terminato un'ora prima che l'ispettore entrasse nella stanza a monte. Guardando all'interno del recipiente attraverso il vetro spia, l'ispettore ha osservato alcune goccioline di condensa e ha notato che le superfici non erano lucide. Ciò non ha convinto l'ispettore della "pulizia visibile" del recipiente (Figura 1).
Figura 1 Alcune impronte chimiche (tracce di scolorimento) rilevate sulla superficie interna del fermentatore
Ciò ha portato alla seguente osservazione: “È stato osservato uno scolorimento nel fermentatore (ID apparecchiatura XXXX). Questo specifico problema non era stato valutato dall'azienda. Il fermentatore viene utilizzato per la fermentazione di [nome del prodotto]. [Nota dell'editore: le citazioni sono tratte dal documento normativo risultante.]
Questa osservazione ha sollevato molti interrogativi e ha influito su diverse strategie di controllo. Poiché si era verificata dopo un ciclo di pulizia in cui era stata eseguita un'ispezione visiva sistematica (di routine), ciò ha influenzato la strategia di controllo della pulizia e il programma di manutenzione in atto per garantire che le superfici del recipiente fossero in condizioni accettabili. Questi due programmi non erano allineati sulle modalità di ispezione e gestione dei recipienti. Cosa potrebbe essere più pratico di un'ispezione visiva giornaliera per confermare uno stato accettabile della superficie prima di confermare lo stato visivamente pulito?
L'azienda biofarmaceutica è stata in grado di giustificare come accettabile la presenza di goccioline dopo il completamento del processo CIP, principalmente perché il tempo di mantenimento della pulizia (CHT) era stato convalidato con successo in condizioni di pulizia simili. L'ispettore ha compreso la situazione (il fatto che l'apparecchiatura fosse stata osservata subito dopo il processo CIP) e ha accettato la giustificazione.
Anche per quanto riguardava lo scolorimento della parete del recipiente è stata fornita una spiegazione. Diverse condizioni possono generare lo scolorimento dell'acciaio inossidabile quando questo entra in contatto con prodotti specifici, agenti chimici o effetti termici. Questo è noto come effetto di degradazione atmosferica interna della superficie. La modifica della superficie può cambiare le condizioni dell'ispezione visiva e, a sua volta, influire sulla decisione dell'esaminatore (o dell'utente) in merito alla pulizia visibile della superficie, anche se il produttore dispone di un processo di formazione e qualificazione di alta qualità.
L'osservazione stessa era chiara: lo “scolorimento […] non era stato valutato” mentre l'apparecchiatura veniva utilizzata regolarmente per produrre massa biologica. Tuttavia, non faceva riferimento ad alcun requisito specifico.
Come ha risposto l'azienda a questa osservazione?
L'azienda ha riconosciuto che durante l'ispezione della FDA è stato rilevato uno scolorimento della superficie, che non era stato valutato prima dell'ispezione.
È stata immediatamente intrapresa un'azione per indagare sullo scolorimento, sono stati prelevati campioni per le analisi e l’utilizzo del fermentatore è stato interrotto.
L'indagine ha dimostrato che la decontaminazione dei recipienti mediante agenti caustici o altre sostanze chimiche ad alte temperature (superiori a 100 °C) sollecitava le pareti del fermentatore e poteva provocare una reazione chimica sulle superfici in acciaio inossidabile. L'elettrolucidatura ripristina solo temporaneamente la superficie dei recipienti. Infatti, i processi di decontaminazione e di pulizia hanno generato nel frattempo degli scolorimenti sulle superfici interne del fermentatore.
Da questa osservazione della FDA possiamo trarre una lezione: l'ambito dell'ispezione visiva effettuata da un esaminatore (ad esempio, utenti o tecnici) dopo un processo di pulizia deve essere ben definito. I processi di decontaminazione e di pulizia hanno un impatto duraturo sulla struttura delle apparecchiature nel tempo. I processi in atto per la pulizia, la decontaminazione e la manutenzione devono essere coerenti, allineati e graduali. Pertanto, la strategia di controllo della pulizia e della manutenzione deve essere in grado di affrontare le seguenti problematiche:
come valutare e documentare le “imperfezioni visive” del materiale che costituisce l’apparecchiatura;
come definire adeguatamente un livello di asciugatura accettabile prima dell'ispezione visiva;
come formare gli esaminatori (utenti, tecnici ed esperti) in materia di pulizia visibile e a riconoscere le imperfezioni superficiali accettabili;
come definire un'imperfezione superficiale che potrebbe portare alla squalifica dell'apparecchiatura;
come stabilire se il ruolo dell'ispezione visiva debba includere il controllo delle imperfezioni superficiali.
Come verificare lo stato "visivamente pulito"
La maggior parte degli esaminatori (utenti) del settore hanno ispezionato e approvato le apparecchiature di produzione basandosi sull'esperienza (1,2). Tuttavia, per gli utenti potrebbe risultare difficile distinguere i residui di prodotti e le imperfezioni superficiali.
Per affrontare questo problema, gli utenti dovrebbero essere formati all'uso di apparecchiature costruite con gli stessi materiali e nelle stesse situazioni in cui si presentano i residui interessati che dovranno rilevare visivamente. La procedura di formazione e il supporto per garantire un'ispezione visiva riproducibile dovrebbero contenere almeno le seguenti sezioni:
1 Definizione di criteri di pulizia visibile
Pulizia visibile, definita come "assenza di residui visibili su una superficie", dovrebbe essere correttamente definita come criterio di accettazione nella procedura.
Oltre alle descrizioni, l'opzione migliore è rappresentata dall'uso di fotografie (Figura 2) di superfici visivamente pulite durante una procedura o un corso di formazione. L'ispezione visiva potrebbe rilevare molto più di semplici residui di prodotto.
Studi recenti hanno dimostrato che formare l'esaminatore a distinguere tra residui sulla superficie e imperfezioni o scolorimenti superficiali non critici è importante per ispezionare visivamente le superfici pulite (2, 3). In alcuni casi, gli utenti potrebbero essere informati del livello di scolorimento o di imperfezione accettabile sulle superfici, poiché alcuni considerano questo controllo della superficie una parte del programma di manutenzione o di ingegneria.
Il livello di asciugatura accettabile dell'apparecchiatura o la categoria e le condizioni di drenaggio potrebbero essere inclusi nella definizione o in una valutazione del rischio di pulizia.
Figura 2 Superfici visivamente pulite dopo un processo di pulizia
2 Ambito dell'ispezione visiva
Quali superfici devono essere sottoposte a ispezione visiva? Tutte le superfici delle apparecchiature che entrano in contatto diretto o indiretto con il prodotto, il materiale sfuso o il materiale intermedio devono essere ispezionate visivamente. Poiché non tutte le superfici di un'apparecchiatura possono essere idonee ad essere sottoposte a ispezione visiva, l'ambito può essere adattato alla sua tipologia specifica. Valutare come e cosa ispezionare visivamente sulla superficie.
3 Condizioni circostanti per ispezionare visivamente una superficie pulita
Le condizioni ambientali in cui si trova l'apparecchiatura da ispezionare visivamente influiscono sulla capacità di rilevare correttamente i residui, rendendo tale ambiente un elemento importante. La quantità di residuo rilevabile (limite di residuo visibile) dipende dal prodotto e deve essere definita con un approccio caso per caso o per gruppi (2–6).
Per confermare l'impatto delle condizioni ambientali su un'ispezione visiva, è necessario analizzare i seguenti fattori:
Distanza di visione: la distanza tra la superficie e l'utente. Questa distanza di visione potrebbe non essere semplice da simulare in laboratorio; tuttavia, il buon senso suggerisce che maggiore è la distanza, più difficile possa essere l'ispezione visiva. Anche l'acuità visiva dell'esaminatore a una distanza specifica gioca un ruolo.
Livelli di luce (circa da 400 a 1500 Lux nelle normali camere bianche): alcuni studi condotti su provini hanno evidenziato che livelli di luce compresi tra 200 e 1400 Lux potrebbero non alterare l'ispezione visiva. Ciò dovrebbe tuttavia essere dimostrato tramite situazioni reali.
Angolo di visione: anche l'angolo di visione tra gli occhi dell'esaminatore e le superfici da ispezionare visivamente dovrebbe essere preso in considerazione e integrato nel processo di ispezione visiva. A seconda dell'angolazione e dell'intensità della luce, un esaminatore potrebbe notare dei riflessi che renderebbero difficile confermare la pulizia visibile.
Luce secondaria: si sconsiglia l'uso di una torcia elettrica quando le sessioni di convalida della pulizia non sono state ispezionate con una torcia elettrica. Le condizioni di ispezione devono essere simili tra le sessioni di convalida e le operazioni di pulizia e manutenzione di routine.
Per evitare di prendere decisioni inadeguate è fondamentale comprendere i fattori circostanti che possono alterare l'ispezione visiva.
4 Tempistiche di esecuzione dell'ispezione visiva
Al termine di ogni operazione di pulizia sarebbe opportuno effettuare un'ispezione visiva, la quale tuttavia spesso non fa parte della documentazione e può dare origine a domande sull'esecuzione dell'ispezione visiva in caso di interruzione di un ciclo di pulizia. La procedura dovrebbe specificare dopo quanto tempo dalla fine del ciclo di pulizia debba essere effettuata un'ispezione visiva. Questa tempistica deve essere determinata per garantire la sicurezza dell'utente durante l'apertura di un recipiente (ad esempio, la fase di risciacquo viene generalmente eseguita ad alte temperature). Questa tempistica influisce sul processo di scarico dell'acqua o del solvente e potrebbe invalidare prematuramente l'ispezione.
5 Metodologia per l'ispezione visiva
Dovrebbe essere stabilita una metodologia strutturata per l'ispezione visiva delle apparecchiature pulite. Ad esempio, un produttore potrebbe sviluppare una lista di controllo che identifichi le varie parti dell'apparecchiatura da ispezionare visivamente in un percorso visivo coerente. Oltre a seguire il metodo dell'ispezione visiva generale, alcuni produttori hanno sviluppato delle liste di controllo in cui vengono identificate parti specifiche o posizioni note come punti critici da ispezionare visivamente.
6 Identificazione dei residui visibili interessati
Agli utenti devono essere presentati tutti gli elementi che potrebbero esaminare durante l'ispezione visiva e che possono includere, tra gli altri, residui di prodotto, imperfezioni superficiali, danni superficiali, residui d'acqua e particelle.
L'utente ispezionerà le superfici visibili con tutte le loro imperfezioni. Sarà inoltre in grado di rilevare molti altri elementi o anomalie, a partire da difetti legati al materiale, al suo invecchiamento, a tracce di acqua residua, particelle e residui di prodotto (Figura 3). Pertanto, l'obiettivo dell'ispezione visiva e i criteri di accettazione devono essere chiaramente definiti.
Figura 3 Elementi che gli utenti devono rilevare su una superficie di contatto diretto o indiretto con il prodotto
Il livello di formalità nello sviluppo di queste sei sezioni dipende dalle strategie di valutazione del rischio e di controllo delle pulizie adottate dall'azienda.
Conclusione
L'ispezione visiva è il metodo preferito per confermare lo stato di pulizia visibile. La maggior parte degli esaminatori (utenti) del settore hanno ispezionato e approvato le apparecchiature di produzione basandosi sull'esperienza (1,3). Tuttavia, potrebbe risultare difficile per gli utenti distinguere i residui dei prodotti e le imperfezioni della superficie senza un'adeguata formazione e qualificazione. Di conseguenza, è sensato formare adeguatamente gli utenti e accertarsi che vengano forniti per l'ispezione tutti i materiali di costruzione e i materiali impiegati in situazioni simili per poter rilevare visivamente i residui interessati e le imperfezioni delle superfici che potrebbero influire sullo stato di pulizia visibile. Dovrebbe essere istituito un programma formale di formazione e qualificazione per l'ispezione visiva, nonché le relative istruzioni. Questo programma dovrebbe basarsi sulla serie di controlli utilizzati dal produttore per verificare la pulizia dell'apparecchiatura. L'ispezione visiva così come definita e qualificata, pertanto, potrebbe rappresentare un metodo di controllo preliminare per una strategia di gestione del rischio di pulizia.
Entrambi gli autori hanno partecipato alla stesura del manoscritto e ne hanno approvato la versione finale. Lo sviluppo di questo articolo è stato sponsorizzato da GlaxoSmithKline Biologicals SA e STERIS. Gli autori dichiarano il seguente conflitto d'interesse: Walid El Azab è un dipendente di STERIS, Stephane Cousin è un dipendente del Gruppo GSK.
Riferimenti parte 1
European Commission, Good Manufacturing Practice Medicinal Products for Human and Veterinary Use: Annex 15, qualification and validation, 2015.
Canada Health Products and Food Branch Inspectorate. Guidance Document. Cleaning validation guidelines: Drug and health products. Health Canada: Ottawa, Canada; 2002 Spring.
European Medicines Agency, Questions and answers on implementation of risk-based prevention of cross-contamination in production and ‘Guideline on setting health-based exposure limits for use in risk identification in the manufacture of different medicinal products in shared facilities’ (EMA/CHMP/CVMP/SWP/169430/2012), EMA/CHMP/CVMP/SWP/246844/2018, (April 2018)
European GMP part IV Guidelines on Good Manufacturing Practice specific to Advanced Therapy Medicinal Products
Pharmaceutical Inspection Convention and Pharmaceutical Inspection Co-operation Scheme. Guide to Good Manufacturing Practice for Medicinal Products: Annex 15, 2015.
Walsh A. et al., “Justification & Qualification Of Visual Inspection For Cleaning Validation In A Low-Risk, Multiproduct Facility.” Pharmaceutical Online (Aug. 3, 2018) accessed January 2020 https://www.pharmaceuticalonline.com/doc/justification-qualification-of-visual-inspection-for-cleaning-validation-in-a-low-risk-multiproduct-facility-0001
ISPE, ISPE Risk-Based Manufacture of Pharmaceutical Products, second edition, Volume 7
Active Pharmaceutical Ingredients Committee, guidance on aspects of cleaning validation in active pharmaceutical ingredient plants (2016)
Parenteral Drug Association, Technical Report 29, Points to Consider for Cleaning Validation (2012)
GUIDE TO INSPECTIONS VALIDATION OF CLEANING PROCESSES, accessed on September 2019: https://www.fda.gov/inspections-compliance-enforcement-and-criminal-investigations/inspection-guides/validation-cleaning-processes-793
Parenteral Drug Association, Technical Report 49, Points to Consider for Biotechnology Cleaning Validation (2010)
ASTM: E306-18, Standard guide for Science-Based and Risk-Based Cleaning Process Development and Validation, October 2018 version 1.
Forsyth, R.J., et al. “Visible-Residue Limit for Cleaning Validation and its Potential Application in a Pharmaceutical Research Facility.” Pharmaceutical Technology 28 (Oct. 1, 2004) 68–72.
“Application of Visible-Residue Limit for Cleaning Validation.” Pharmaceutical Technology 28 (Oct. 2, 2005) 10 http://www.pharmtech.com/application-visible-residue-limit-cleaning-valiation?id=&pageID=1&sk=&date= (accessed September 2019)
“Determination of Surface Visible Residue Limits on Pharmaceutical Plant Equipment.” Pharmaceutical Technology 37 (Feb. 2, 2013)
Desai, P., and Walsh, A. “Validation of Visual Inspection As An Analytical Method For Cleaning Validation.” Pharmaceutical Online (Sept. 11, 2017) https://www.pharmaceuticalonline.com/doc/validation-of-visual-inspection-as-an-analytical-method-for-cleaning-validation-0001 (accessed September 2019)
International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for human use, Quality Risk Management Q9, (2005)
Walsh, A., et al., “Justification & Qualification Of Visual Inspection For Cleaning Validation In A Low-Risk, Multiproduct Facility.” Pharmaceutical Online (Aug. 3, 2018) https://www.pharmaceuticalonline.com/doc/justification-qualification-of-visual-inspection-for-cleaning-validation-in-a-low-risk-multiproduct-facility-0001 (accessed January 2020)
Forsyth, R.J., and Hartman, J., “A Risk-based Approach to Cleaning Validation using Visible Residue Limits.” Pharmaceutical Engineering 28 (2008) 8–22.
Riferimenti parte 2
El Azab W. and Cousin S., “Visual Inspection Practices of Cleaned Equipment: Part I,” accessed April 2020: https://www.pda.org/pda-letter-portal/home/full-article/visual-inspection-practices-of-cleaned-equipment-part-i
Walsh, A., et al., “Justification & Qualification of Visual Inspection for Cleaning Validation in a Low-Risk, Multiproduct Facility.” Pharmaceutical Online (Aug. 3, 2018) https://www.pharmaceuticalonline.com/doc/justification-qualification-of-visual-inspection-for-cleaning-validation-in-a-low-risk-multiproduct-facility-0001
Desai, P. and Walsh, A. “Validation of Visual Inspection as an Analytical Method for Cleaning Validation.” Pharmaceutical Online (Sept. 11, 2017) https://www.pharmaceuticalonline.com/doc/validation-of-visual-inspection-as-an-analytical-method-for-cleaning-validation-0001
Forsyth, R.J., et al. “Visible-Residue Limit for Cleaning Validation and its Potential Application in a Pharmaceutical Research Facility.” Pharmaceutical Technology 28 (Oct. 2004): 58–72 https://pdfs.semanticscholar.org/ea64/d01db84d3b8a5a62f1c824abf8484af363dd.pdf
“Application of Visible-Residue Limit for Cleaning Validation.” Pharmaceutical Technology 29 (Oct. 2, 2005) http://www.pharmtech.com/application-visible-residue-limit-cleaning-validation?id=&pageID=1&sk=&date=
“Determination of Surface Visible Residue Limits on Pharmaceutical Plant Equipment,” Pharmaceutical Technology (Feb. 2, 2013) 37 http://www.pharmtech.com/determination-surface-visible-residue-limits-pharmaceutical-plant-equipment?id=&pageID=1&sk=&date=
Contenuti consigliati
Articolo
Un processo giustificato per la pulizia e la disinfezione
Le linee guida e gli standard normativi, come lo European Good Manufacturing Practice (UE GMP) Annex 1, vengono creati per fornire standard per i requisiti. Sebbene possa essere utile cercare di attenersi alle migliori pratiche, è anche importante ricordare che le procedure devono essere adattate alle proprie strutture e processi aziendali. In questo articolo Walid El Azab, Technical Expert per STERIS, affronta l'approccio scientifico per determinare se è necessaria una fase di pulizia separata prima della disinfezione.
Servizi da tenere in considerazione nella convalida delle apparecchiature
La convalida costituisce una parte fondamentale della propria attività. Assicurati che le apparecchiature siano conformi agli standard e alle normative di settore grazie ai servizi di convalida di STERIS. Che si tratti di prima qualifica o di qualifica periodica annuale, STERIS offre un pacchetto di servizi in grado di soddisfare le tue esigenze in ogni fase del processo.
Per maggiori dettagli sui pacchetti di servizi disponibili, consulta l'infografica.
Preservazione dell'acciaio inossidabile: mitigazione del rouging
Questo white paper esamina le migliori pratiche per la definizione e la valutazione di un processo di conservazione e mitigazione delle macchie rosse dell'acciaio inossidabile nella produzione farmaceutica.
Un processo giustificato per la pulizia e la disinfezione
Le linee guida e gli standard normativi, come lo European Good Manufacturing Practice (UE GMP) Annex 1, vengono creati per fornire standard per i requisiti. Sebbene possa essere utile cercare di attenersi alle migliori pratiche, è anche importante ricordare che le procedure devono essere adattate alle proprie strutture e processi aziendali. In questo articolo Walid El Azab, Technical Expert per STERIS, affronta l'approccio scientifico per determinare se è necessaria una fase di pulizia separata prima della disinfezione.
Servizi da tenere in considerazione nella convalida delle apparecchiature
La convalida costituisce una parte fondamentale della propria attività. Assicurati che le apparecchiature siano conformi agli standard e alle normative di settore grazie ai servizi di convalida di STERIS. Che si tratti di prima qualifica o di qualifica periodica annuale, STERIS offre un pacchetto di servizi in grado di soddisfare le tue esigenze in ogni fase del processo.
Per maggiori dettagli sui pacchetti di servizi disponibili, consulta l'infografica.
Preservazione dell'acciaio inossidabile: mitigazione del rouging
Questo white paper esamina le migliori pratiche per la definizione e la valutazione di un processo di conservazione e mitigazione delle macchie rosse dell'acciaio inossidabile nella produzione farmaceutica.